hidden
Over 10 years experience of Traceability Solutions
By Pharmatrax Author
Category: Technoloy
 No Comments
No Comments
Cancer Drug Discovery’s “RASzle-Dazzle”
Despite decades of research and therapeutic advances, cancer remains a global scourge and is currently the second leading cause of mortality. According to the American Association for Cancer Research (AACR), in 2018, there were an estimated 17 million new cases worldwide with 9.5 million deaths. By 2040, those numbers are expected to climb to 27.5 million new cases and 16.3 million deaths (largely due to aging populations).
Despite these gloomy statistics, researchers also have made steady progress in developing anticancer therapeutics. The AACR reported that the overall U.S. cancer death rate declined by 27% from 1991 to 2016, saving more than 2.6 million lives. Further, in the period August 1, 2018 to July 31, 2019, the FDA approved 17 new anticancer therapeutics and expanded the indications of 10 approved anticancer therapeutics to additional types of cancer.
New strategies for developing anticancer therapeutics are featured in the program for the Cambridge Healthtech Institute’s 15th annual Drug Discovery Chemistry meeting. Scheduled to take place August 24–28 in San Diego, the meeting will, if all goes to plan, be an opportunity to hear presentations on strategies such as the targeting of specific cancer-causing mutations in RAS genes, the modulation of protein secretion, and the identification of compounds capable of targeting transcription to attack cancer cell vulnerabilities.
Selected presentations are previewed in this article, which also highlights one of the meeting’s featured presentations, a lecture describing how research collaborations culminated in the development of a pan-FGFR inhibitor. The lecture is expected to emphasize that drug development can be accelerated if scientists representing academia, biotech, and the pharmaceutical industry embrace a team approach.
Targeting aberrant RAS proteins
Mutations in RAS genes drive some of the deadliest cancers. Representing the “dark side” of RAS, these mutations lead to the expression of malfunctioning RAS proteins, GTPases that control cytoplasmic signaling networks involved in cellular growth, differentiation, and survival. According to the National Cancer Institute, more than 30% of all human cancers (including 95% of pancreatic cancers and 45% of colorectal cancers) result from mutations in RAS proteins. The RAS protein family comprises three major isoforms (KRAS, NRAS, and HRAS). Of these isoforms, it is KRAS that is most often associated with cancer. In fact, KRAS alterations account for up to 87% of cancer-causing RAS mutations.

Druggable targets for RAS-mediated cancers have been elusive, despite 30 years of intense pursuit. The National Cancer Institute’s RAS Initiative, established in 2013 and headed by Frank McCormick, PhD, FRS, is mobilizing government, academic, and industry researchers to change that picture.
Serving as the RAS Initiative’s reagents group lead is Dominic Esposito, PhD, the director of the Protein Expression Laboratory at the Frederick National Laboratory for Cancer Research. He reports, “Overall, we want to better understand RAS biology, especially at the plasma membrane, and secondly, to coordinate the screening of new drugs.”
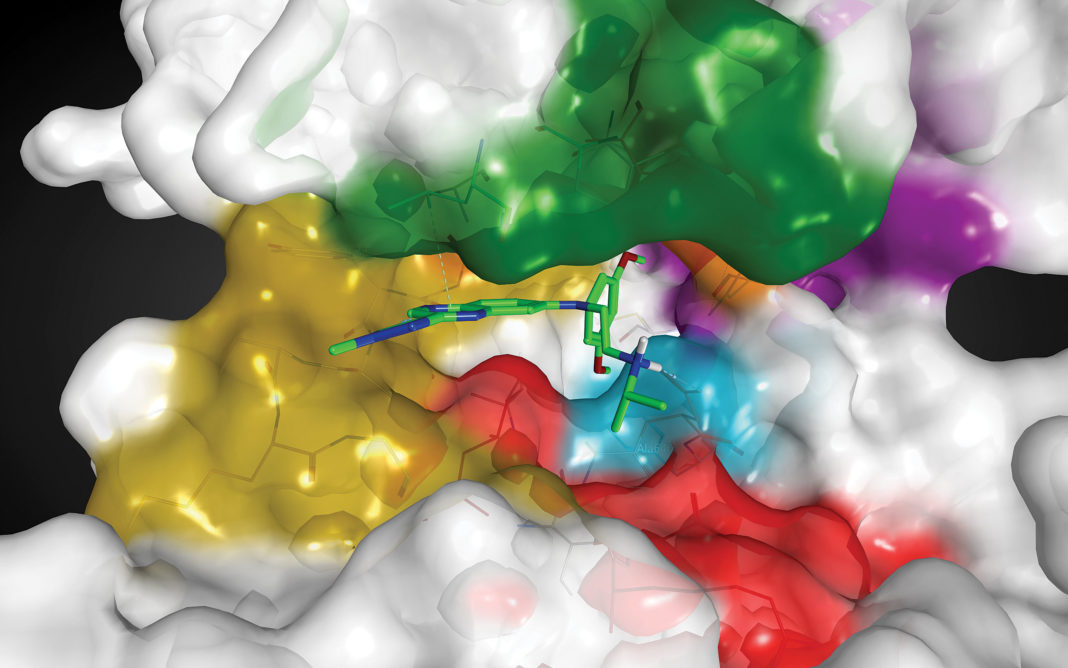
Novel approaches are needed since RAS proteins lack typical “pockets” important for therapeutic targeting while also possessing high specificity and affinity for their common substrate, GTP. “A number of promising drug discovery efforts have failed after preclinical work,” explains Esposito. “Part of the problem was that investigators often utilized a non-membrane part of RAS called the G domain. We have developed a high-yield process for producing the accurate membrane-bound form of RAS, and biochemical, biophysical, and cell-based assays to provide a larger toolbox.”
According to Esposito, one approach utilizes a “covalent tethering strategy.” He elaborates, “We developed a novel tethering library, and instead of targeting only native cysteines, as others have done, we mutated all surface amino acids of KRAS to cysteine one at a time, searching for a novel pocket or handle we could target.”
Although initially any hits obtained by these tethering screens would be low affinity, the ultimate goal is to improve such compounds into clinical candidates. Esposito clarifies, “Once we identify fragment hits, we can combine computational modeling and structure-guided approaches to potentially reach outside the pocket to increase binding affinity and to produce more drug-like candidates.”
Ongoing clinical trials
KRAS G12C, one of the most frequent KRAS mutations, locks the RAS protein in an activated state leading to increased cellular signaling and unchecked tumor growth. This mutation drives cancers such as non-small-cell lung cancer (NSCLC), colorectal adenocarcinomas, and pancreatic cancers.
Targeted therapies to treat such cancers are being developed by Mirati Therapeutics. The company’s unique small-molecule inhibitor, MRTX849, was created by utilizing a structure-based drug design approach and optimization for favorable drug-like properties. This selective and covalent inhibitor irreversibly turns the tables by locking the KRAS G12C protein in its inactive state. Consequently, tumors shrink and die. MRTX849 is among the first such inhibitors to advance to clinical trials.
James G. Christensen, PhD, Mirati’s chief scientific officer, is leading a collaborative effort assessing the antitumor activity of MRTX849 in vitro and in vivo. The group recently published a paper comprehensively detailing the molecular characterization of MRTX849, its effects on multiple tumor models, and the correlation with baseline and dynamic molecular response to KRAS inhibition. The team’s studies employed human KRAS G12C-positive lung and pancreatic cell lines as well as a panel of mutant cell line–derived xenografts and patient-derived xenograft models. According to Christensen, these studies identified mechanisms involved in limiting antitumor activity including KRAS-nucleotide cycling and pathways inducing feedback reactivation and/or bypassing KRAS dependence.
Studies were also performed that expanded into the functional genomics arena including CRISPR and combination approaches. They found that regulatory nodes were able to sensitize tumors to KRAS inhibition when co-targeted. Further, studies combining targeted therapies with MRTX849 treatment suggested that virtually all KRAS G12C-mutated cancers can derive clinical benefit from either direct KRAS inhibitor therapy alone or in combination with other drugs. Christensen believes the discovery of MRTX849 provides a long-awaited opportunity to selectively target KRAS G12C in patients. Mirati is evaluating MRTX849 in a clinical trial and will initiate additional trials using MRTX849 as a single agent or in combination therapy.
Next-generation medicinal chemistry
Most secreted and transmembrane proteins undergo post-translational processing in the endoplasmic reticulum (ER) to add appropriate sugars or to facilitate proper folding prior to extracellular or membrane targeting. One of the key components facilitating insertion into the ER is a channel called Sec61. As part of a collection of proteins referred to as the translocon, Sec61 interacts with unique signal sequences of growing polypeptide chains thereby permitting them across the ER membrane. Several protein clients of Sec61 are of therapeutic interest, including cancer growth factors and receptors.
Kezar Life Sciences is striving to modulate protein secretion at the Sec61 level. “This is a first-in-class approach to cancer treatment,” asserts Dustin McMinn, PhD, the company’s senior director and head of chemistry. “The concept we propose targets protein homeostasis. Almost all secreted and transmembrane proteins utilize Sec61 as a gateway toward functionalization. Although there already are a number of compounds that broadly block Sec61, most target too aggressively, leading to toxicity and/or poor drug-like properties.”
He continues, “We wanted to develop drug-like protein secretion inhibitors that can safely target cancer. To do this, we produced small molecules that block the functional interaction of signal sequences and Sec61. With now multiple classes of such compounds, we can inhibit secretion of a specific protein or group of proteins.”
The company’s preclinical studies utilized in vitro protein secretion assays and found a candidate, KZR-261, that blocks expression of therapeutically relevant targets and inhibits growth of multiple in vivo tumor xenografts.
McMinn explains, “Utilizing this approach, we inhibit protein translocation with improved tolerability. KZR-261 can induce simultaneous expression inhibition of multiple, clinically relevant proteins including oncogenic drivers, angiogenic factors, and immune checkpoints.”
The company has initiated Investigational New Drug (IND)-enabling studies for intravenously administered KZR-261. The IND filing, which is expected in the first quarter of 2021, may take KZR-261 to the next stage of development, namely, evaluation of the drug against multiple solid tumor types.
CDKs as transcriptional targets
Cyclin-dependent kinases (CDKs) that control cell proliferation have long attracted the eye of researchers developing anticancer drug targets. However, CDKs also regulate RNA polymerase II–dependent transcription cycles. Transcriptional CDKs have recently emerged as potential anticancer therapeutic targets, but a deeper understanding of their functions is needed to drive their therapeutic potential.
According to Robert P. Fisher, MD, PhD, professor of the Department of Oncological Sciences, Icahn School of Medicine at Mount Sinai, targeting CDKs in transcription machinery has proved challenging since they are active in dividing as well as nondividing cells. However, not all Pol II–mediated transcription is equally dependent on CDKs. Characterization of tumor-specific super-enhancers (SEs) revealed a potential vulnerability of cancer cells to drugs targeting transcription.
Fisher notes that CDK7 is enriched in SEs, regulatory DNA elements that recruit enhanced levels of transcription machinery to drive high-level expression of genes that determine cell identity and survival. He further indicates that disruption of SE-dependent transcription is the likely basis for the tumor-specific cytotoxic effects in preclinical studies of THZ1, a promising covalent CDK inhibitor with antitumor activity for many types of cancers.
However, the greatest anticancer potential of CDK therapeutics may be in their ability to be utilized in combination therapies. For example, the combination of CDK7 inhibition with p53 activation is synthetically lethal to susceptible cancer cells. Synthetic lethality refers to a lethal effect resulting from perturbation of two genes simultaneously to produce loss of viability, whereas a cell remains viable after the perturbation of either gene alone.
The exploration of CDK mechanisms is continuing apace, as is the development of CDK therapeutics that inhibit transcription. Nonetheless, many questions remain. For example: Do CDKs act alone or are other elements involved? Will tumors be differentially susceptible to drugs or drug combinations targeting CDKs?
Fisher believes answers will emerge over the next decade with improved structural and biochemical insights into transcriptional processes and with the emergence of powerful chemical, genetic, and genomic tools to reveal the needed insights. He projects, “Ongoing studies will lead to a better understanding of the fundamental processes controlled by CDKs, and of the consequences of perturbing those processes in both cancer cells and normal tissues. Moreover, these investigations should reveal new network components, such as proteins modified by CDKs to drive downstream effects on transcription, or phosphatases—the enzymes that reverse those modifications—that might also be promising anticancer drug targets.”
The team approach
The concept that the sum is greater than its parts applies to the robust synergy that can develop when pharma, academia, and biotech entities pool their resources to tackle cancer drug discovery. Case in point: A remarkable collaboration of three such entities led to the discovery of erdafitinib (Balversa™), which inhibits the four fibroblast growth factor receptor (FGFR) kinases. In 2019, erdafitinib became the first FGFR-targeted drug to be approved by the FDA. In fact, the FDA granted accelerated approval following Phase II studies demonstrating that the drug has a 32% response rate for locally advanced or metastatic urothelial carcinoma patients whose tumors possessed actionable FGFR alterations and who previously received platinum-based chemotherapy.
The Northern Institute of Cancer Research at Newcastle University worked with Astex Pharmaceuticals to identify early FGFR inhibitors. Subsequently, Astex, in
collaboration with Janssen Pharmaceutica, optimized and clinically developed small-molecule inhibitors of FGFR. The teams toiled ~11 years fine-tuning potencies and ligand efficiency via medicinal chemistry optimization and progressing through preclinical in vivo work to clinical trials.
The collaborators utilized a fragment-based approach, which has become a powerful tool not only for discovering drug leads but also to reduce off-target effects and enhance selectivity. The process begins with very small compounds whose tiny size results in far fewer possible fragments as compared to the much larger drug-like molecules identified by high-throughput screening approaches.
Patrick Angibaud, PhD, scientific director and fellow, Internal R&D Discovery, Janssen, reflects, “Collaboration and open innovation among multiple teams led to the discovery of erdafitinib, with each team bringing forth its specific expertise.” According to Angibaud, such a collaborative model involves academic and biotech partners 1) cultivating biological knowledge and 2) taking advantage of pharmaceutical companies’ knowledge and tools for compound optimization as well as expertise in preclinical and clinical development.
source : https://www.genengnews.com/insights/cancer-drug-discoverys-raszle-dazzle/


